Sequencing Pathogens from Clinical Samples
Pathogen sequencing has developed, in the past decades, from its origin in the characterisation of bacterial and viral isolates prepared using time-honoured techniques of microbiology. Early papers on using genomics to investigate outbreaks, such as Lewis et al’s 2010 paper on an A. baumannii outbreak in a British hospital and Rico-Hesse et al’s 1987 paper describing poliovirus 1 genotypes purified DNA and RNA from cultures prepared from clinical isolates. Sequencing from culture has several disadvantages: pathogen culture requires a laboratory and workforce trained in biosafety, takes time (especially for slow-growing microbes) and might introduce bias through the impact of selective pressure on pathogens (especially fast evolving ones like viruses).
Directly sequencing from clinical samples poses its own challenges, foremost of which is the need to distinguish the pathogen genetic sequence from host sequence and other contaminants. There are several approaches to address this challenge:
-
Metagenomic sequencing, sometimes after host depletion through methods like differential centrifugation and (for bacteria) differential lysis.
-
Hybrid capture using RNA-bait coated beads
-
Tiled-amplicon sequencing
Metagenomic (i.e. whole sample) sequencing typically has low yields of pathogen sequence, limiting the ability to multiplex samples in a single sequencing run and thereby increasing the cost per sample. While effective at capturing pathogen genetic material, hybrid capture approaches are more expensive than tiled-amplicon sequencing for small pathogens such as SARS-CoV-2.
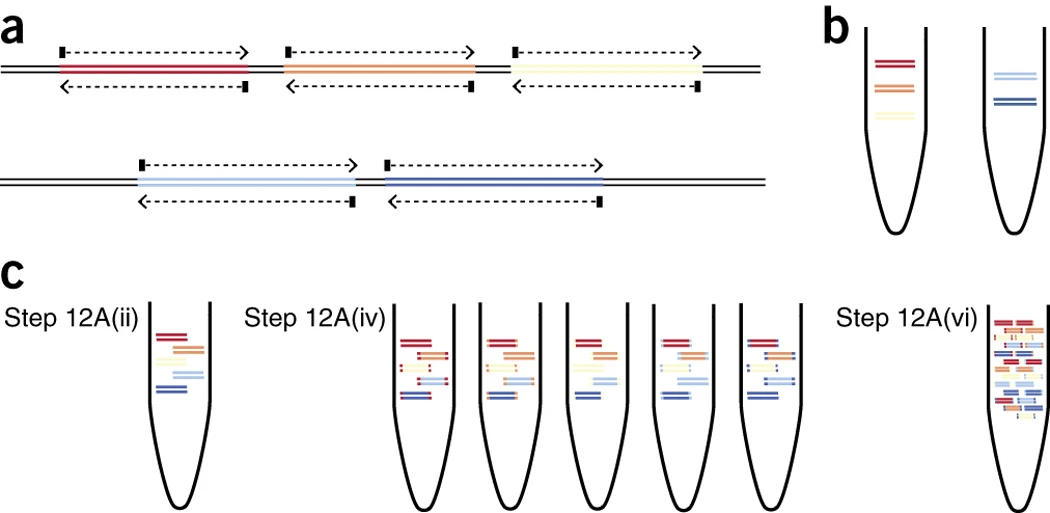
The tiled-amplicon approach to sequencing viruses was first popularised by the ARTIC Network. In their 2017 paper, Josh Quick et al describe a method known as Primal Scheme for generating sequencing primers from viral genomes. After choosing an amplicon size, left and right primers are designed for use in two primer pools. The amplicons in each pool are non-overlapping but when the amplification products from both pools are combined they provide coverage of the complete genome.
Challenges of the ARTIC amplicon protocol
Characteristics of the SARS-CoV-2 virus and its replication pose several challenges for sequencing using the ARTIC amplification protocol:
Firstly, the SARS-CoV-2 genome is replicated both as a whole and as several subgenomic RNAs. As a result, the concentration of genomic material in a sample might not evenly cover the virus genome, resulting in changing coverage across the viral genome. Attempts to address this have included generating alternative primer schemes and also through changing the concentration (balancing) of different primers in the primer pools.
Secondly, mutations in the virus can affect the primer binding sites for amplicons. This can lead to “amplicon dropout” where the region covered by an amplicon sequence is only present at very low coverage or not at all. Mistakes in primer design can also lead to unintended primer interaction and also contribute to amplicon dropout. As multiple viruses from diverse lineages are sometimes handled in the same lineage, amplicon dropout can also increase the risk of contamination and spurious results, as in the case of apparent “Deltacron” sequences.
Primer Schemes and Bioinformatic processing
As mentioned above, amplicons are produced in the ARTIC amplicon protocol through amplification of genomic sequence using PCR primers. The ends of these amplicons are thus comprised of sequence from the sequencing primers themselves. While long read sequencing, when using certain library preparation protocols, will capture primers at the end of reads, short read sequencing and tagmentation based protocols will result in reads where the parts that are made up of primers are not clear. Each amplicon sequencing kit thus is accompanied by a description of the position of primers on the viral genome. With this description in hand, bioinformatic techniques can trim (i.e. remove) the primer bases from reads after the reads are aligned against a viral genome and thus the genomic orientation of the read and the read orientation are known.
Certain forms of amplicon dropout and primer problems can lead to primer-derived bases being missed (and thus create an apparent reversion to wild-type genome). This problem can be addressed by requiring that reads do not span amplicon boundaries, but its existence (and discussion in open forums) illustrates the importance for community efforts in spotting and debugging unexpected problems as pathogens, sequencing protocols and bioinformatics approaches evolve.
Up until now there have been several inconsistent mechanisms for publishing information on which primers are used in amplicon sequencing protocols and the primer positions. A project is, however, underway within PHA4GE to standardise the format for publishing primer schemes, the results of which can be seen in this Github repository. This effort aims to both standardise how primer schemes are published and also how they are mentioned in descriptions of methods and in databases. For example, it is not sufficient to note that sequencing was done with “tiled amplcons” or even “ARTIC amplicons” as the version and thus details of a the primer scheme used are necessary for bioinformatics analysis.
In addition to the various versions of the ARTIC SARS-CoV-2 primer protocols, the PHA4GE repository has captured information on the evolution of the “Midnight primers”. Original developed by Nikki Freed, Olin Silander and colleagues, the Midnight scheme uses 1200 base pair amplicons (in contrast to the ARTIC network’s 400 base pair amplicons).
Midnight primers were adopted by Oxford Nanopore in their “Midnight kits” but also independently evolved both by the original authors and by John Tyson and others at the British Columbia Centre for Disease Control (BC-CDC) in Canada. There are thus several, slightly divergent, versions of the Midnight scheme currently in use.
Sampling from clinical samples - beyond SARS-CoV-2
Tiled amplicon primers schemes have been developed for several other viruses, such as Zika, Ebolavirus Zaire, Mpox virus (MPXV), Dengue fever virus, Measles virus and more. The requirement to cover the entire genome and for a close match between primer and virus genome means that primer schemes need often need to be designed with a specifical viral strain in mind and also that schemes can get large - the MPXV scheme from Vogels et al contains 163 amplicons, each 2000 base pairs long.
Tiled amplification has thus not been used for sequencing of bacteria from clinical samples. Some of the other techniques mentioned above, including laboratory-based “host depletion” and RNA-bait hybrid capture have been used for bacteria and parasites. Other approaches to sequencing from clinical samples have included targetted sequencing (for example for the “drug resistance” genes of M. tuberculosis) and amplification using Selective Whole Genome Amplification (SWGA), a multiple-displacement amplification technique that has been used for Plasmodium species (see 1 and 2) as well as Leishmania and bacterial species such as T. pallidum and N. meningitidis.
Sequencing from clinical samples continues to be an active area of research in sequencing and bioinformatics and skills learned in applying the technique to one pathogen are readily transferable to others, especially when bioinformatics workflows exist to automate at least some steps of the bioinformatics protocols.